Sunlight is a multi-faced source of energy since it impacts life through intricately woven networks of beneficial as well as harmful biophysical and biochemical transformations. This suggests that the effects of photoexcited electrons on downstream processes is subtle and mechanistic insights into such light-induced electronic processes in natural and artificial systems can result in novel ways to utilize sunlight for constructive purposes. Our primary interests lie in understanding light-induced vibronic phenomena, focusing specifically on photovoltaic applications and combating ultraviolet light induced aggregation of biomolecules, using first-principles based theories, mixed quantum-classical and quantum mechanical-molecular mechanics algorithms.
Method development projects include detailed investigation of the localized operator partitioning method (LOPM-CIS, LOPM-TDDFT) and implementation of open-shell wavefunction based overlap method (OD-scheme) for calculating time-dependent nonadiabatic couplings in the Newton-X program.
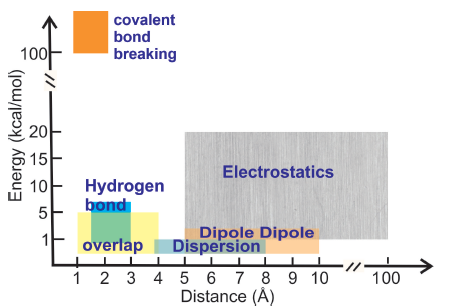
Cooperativity of weak noncovalent interactions in biology
Noncovalent interactions (NCIs) such as pi-pi, cation-pi, anion-pi, XH-pi(X=N,C,O) and lone pair-pi have recently gained importance in biology due to their cooperative role in the self-assembly and phase separation of membraneless organelles in cells, protein folding, molecular recognition, drug discovery and development of biomimetic `smart' materials. NCIs are distinct from the well-known electrostatic and van der Waals' interactions ( see Figure on the right). Since NCIs are weak in nature, they are capable of providing much more flexibility in structures compared to the covalent bond and provide energetically cheaper ways of constraining the conformational space of biomolecules to achieve function. Several NCIs are quantum mechanical in nature due to the overlap of electronic cloud distributions (or orbitals) between different atom groups (e.g., pi-pi,lone pair-pi) -- these are not easy to characterize in molecules with >100 atoms such as proteins. However, the advancement of bioinformatics, cheminformatics and quantum chemical techniques have brought forth the possibility of chemical characterization of \glspl{nci} in larger systems in biology. The investigation of NCIs from a quantum perspective is at a nascent stage, and a multi-scale approach is essential to obtain a wholistic understanding of their role in guiding biological structure and function.
One of the most challenging aspects of understanding NCIs in biology is the sheer diversity of short- and long-range overlap based interactions. It has been hypothesized that the synergy between NCIs (multivalency) could be a key principle in driving several biological functions, though the extent of cooperativity between one or more interaction types is only beginning to be investigated. A second challenge is that the interactions can be repulsive or attractive: steric effects are repulsive, and are useful for rationalizing stereocontrol in strongly bound and conformationally restricted complexes. Such complexes do not undergo significant structural reordering because of an impending high energy penalty. On the other hand, NCIs are weaker, less directional and energetically do not change much upon significant structural reorganization. This is the reason why NCIs have to act cooperatively to constrain the conformational space. The overall goal of this project will be to develop an efficient protocol to characterize the role of NCIs that is generalizable to any biological system.
Noncovalent interactions (NCIs) such as pi-pi, cation-pi, anion-pi, XH-pi(X=N,C,O) and lone pair-pi have recently gained importance in biology due to their cooperative role in the self-assembly and phase separation of membraneless organelles in cells, protein folding, molecular recognition, drug discovery and development of biomimetic `smart' materials. NCIs are distinct from the well-known electrostatic and van der Waals' interactions ( see Figure on the right). Since NCIs are weak in nature, they are capable of providing much more flexibility in structures compared to the covalent bond and provide energetically cheaper ways of constraining the conformational space of biomolecules to achieve function. Several NCIs are quantum mechanical in nature due to the overlap of electronic cloud distributions (or orbitals) between different atom groups (e.g., pi-pi,lone pair-pi) -- these are not easy to characterize in molecules with >100 atoms such as proteins. However, the advancement of bioinformatics, cheminformatics and quantum chemical techniques have brought forth the possibility of chemical characterization of \glspl{nci} in larger systems in biology. The investigation of NCIs from a quantum perspective is at a nascent stage, and a multi-scale approach is essential to obtain a wholistic understanding of their role in guiding biological structure and function.
One of the most challenging aspects of understanding NCIs in biology is the sheer diversity of short- and long-range overlap based interactions. It has been hypothesized that the synergy between NCIs (multivalency) could be a key principle in driving several biological functions, though the extent of cooperativity between one or more interaction types is only beginning to be investigated. A second challenge is that the interactions can be repulsive or attractive: steric effects are repulsive, and are useful for rationalizing stereocontrol in strongly bound and conformationally restricted complexes. Such complexes do not undergo significant structural reordering because of an impending high energy penalty. On the other hand, NCIs are weaker, less directional and energetically do not change much upon significant structural reorganization. This is the reason why NCIs have to act cooperatively to constrain the conformational space. The overall goal of this project will be to develop an efficient protocol to characterize the role of NCIs that is generalizable to any biological system.
|
|
Photoprotection in biomolecules
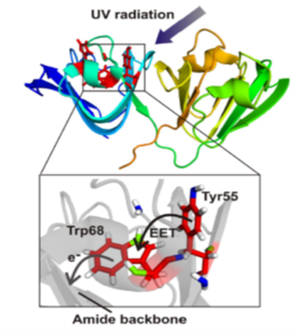
UV-light causes photodegradation of biomolecules resulting for instance in cancerous DNA lesions and cataracts of the eye lens. Though the eye lens cataracts are caused by the aggregation of the crystallin family of proteins from accummulated exposure to UV-light, a remarkable feature of the eye lens is that it possesses protective mechanisms to prevent the UV-damage and maintain the folded conformation of the proteins over nearly the lifetime of an individual. Understanding these mechanisms will be crucial to develop physico-chemical strategies that can repair and reverse the photodamage in cataract.
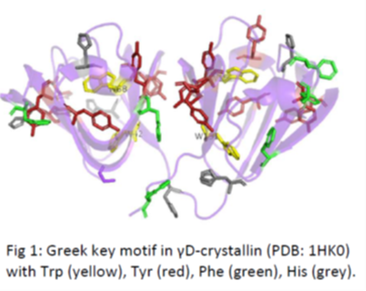
Photoprotective mechanisms often involve aromatic amino acids (above figure on the right), and the canonical approach has been to investigate light induced electronic pathways initiated from Tryptophans because of their highest light absorptivity. However Tryptophans are ten-fold less abundant than the next highest absorbers, namely the Tyrosines, and are also more buried than the Tyrosines. We are interested in investigating photoinitiated pathways from the Tyrosines point of view, and the figure above on the left shows one such pair of Tyrosine-Tryptophan that is being currently investigated.
UV-light causes photodegradation of biomolecules resulting for instance in cancerous DNA lesions and cataracts of the eye lens. Though the eye lens cataracts are caused by the aggregation of the crystallin family of proteins from accummulated exposure to UV-light, a remarkable feature of the eye lens is that it possesses protective mechanisms to prevent the UV-damage and maintain the folded conformation of the proteins over nearly the lifetime of an individual. Understanding these mechanisms will be crucial to develop physico-chemical strategies that can repair and reverse the photodamage in cataract.
Photoprotective mechanisms often involve aromatic amino acids (above figure on the right), and the canonical approach has been to investigate light induced electronic pathways initiated from Tryptophans because of their highest light absorptivity. However Tryptophans are ten-fold less abundant than the next highest absorbers, namely the Tyrosines, and are also more buried than the Tyrosines. We are interested in investigating photoinitiated pathways from the Tyrosines point of view, and the figure above on the left shows one such pair of Tyrosine-Tryptophan that is being currently investigated.
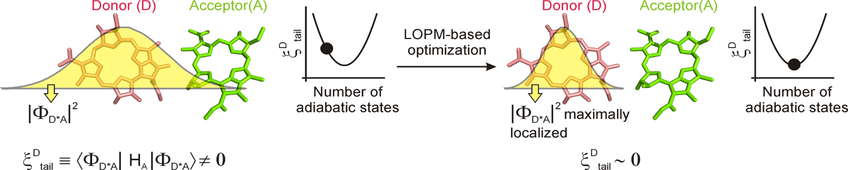
Fragment based methods to construct diabatic states for electronic energy transfer
Chemical intuition is an indispensable guide to rationalize elementary molecular events and has led to the concept of 'diabatic' states. The diabatic states are obtained by a unitary transformation of the adiabatic states resulting from the solution of the time-independent electronic Schrodinger equation within the Born-Oppenheimer approximation. In the context of energy and charge transfer processes, the diabatic states are envisioned to be localized in excitation energy or in charge so as to facilitate the interpretation of the actual transfer of excitation energy or charge from the donor to the acceptor moieties. Practical restrictions in the size of the adiabatic basis makes it difficult to achieve fully excitation-energy localized diabatic basis. Here we develop a tool to construct maximally localized diabatic basis based on fragment excitation energy criteria within the localized operator partitioning method framework. The new tool will help in verifying the actual amount of localization of the excitation energy on a fragment and thereby aid in a robust interpretation of molecular dynamics simulations of the energy transfer phenomenon.
Chemical intuition is an indispensable guide to rationalize elementary molecular events and has led to the concept of 'diabatic' states. The diabatic states are obtained by a unitary transformation of the adiabatic states resulting from the solution of the time-independent electronic Schrodinger equation within the Born-Oppenheimer approximation. In the context of energy and charge transfer processes, the diabatic states are envisioned to be localized in excitation energy or in charge so as to facilitate the interpretation of the actual transfer of excitation energy or charge from the donor to the acceptor moieties. Practical restrictions in the size of the adiabatic basis makes it difficult to achieve fully excitation-energy localized diabatic basis. Here we develop a tool to construct maximally localized diabatic basis based on fragment excitation energy criteria within the localized operator partitioning method framework. The new tool will help in verifying the actual amount of localization of the excitation energy on a fragment and thereby aid in a robust interpretation of molecular dynamics simulations of the energy transfer phenomenon.
